
Привіт Гість ( Вхід | Реєстрація )
 Розподілені обчислення в Україні > Великі проекти розподілених обчислень > World Community Grid > Завершені проекти WCG
Розподілені обчислення в Україні > Великі проекти розподілених обчислень > World Community Grid > Завершені проекти WCG
| Rilian |
 Dec 21 2008, 04:23 Dec 21 2008, 04:23
Пост
#1
|
 interstellar  Група: Team member Повідомлень: 17 377 З нами з: 22-February 06 З: Торонто Користувач №: 184 Стать: НеСкажу Free-DC_CPID Парк машин: ноут и кусок сервера |
 Помоги вылечить мускульную дистрофию, 2 фаза / Help Cure Muscular Dystrophy, Phase 2 Как присоединиться читайте в главном топике World Community Grid 
 Введение В 1986 году был идентифицирован первый ген (dystrophin gene), вовлеченный в дистрофию Дюшена, самую общую форму мышечной дистрофии. Благодаря генетическим исследованиям сегодня известно более 200 генов, вызывающих нервно-мышечные болезни. Однако, до сих пор знания о функциях и взаимодействии между собой белков, закодированных этими генами, остаются крайне незначительными. Раскрытие механизмов возникновения и развития нервно-мышечных заболеваний (патогенез), учитывая большое количество вовлеченных в него генов и кодируемых ими белков, становится все более сложной задачей по мере увеличения наших знаний в этой области. Все клетки содержат одинаковую генетическую информацию в виде ДНК, которая в целом образует геном организма. Процесс экспрессии генов (у человека найдено более 20 тысяч генов), содержащихся в геноме, приводит к образованию белков (более 45 тысяч, т.е. в среднем один ген кодирует 1,6 белков), без которых невозможно функционирование клеток, в том числе специфических, например, мышечных. Некоторые их этих белков являются ферментами, другие - сигнальными молекулами, а могут быть и рецепторами, специфически связывающиеся с другими молекулами (лигандами); а также - структурными белками. Конформация (геометрическая форма, которую принимает органическая молекула) белка в пространстве (3D-структура) определяет его природные функции. Функционирование мышц зависит от многих белков. Эти белки локализованы и работают на различных уровнях - в оболочке клетки, ее протопласте (внутреннем содержимом) или в аксоне двигательного нерва, который соединяет нерв с мышечной тканью и передает команды нервной системы. Большинство нервно-мышечных болезней возникает под действием генетических изменений (мутации в определенных генах вызывают изменения в трехмерной структуре кодируемого ими белка), приводящих к утрате белками полностью или частично их первоначальных функций или к прекращению синтезирования белка как такового. Нервно-мышечные заболевания можно классифицировать по белкам или участкам белков, видоизменившихся под действием мутации генов. Поэтому, чтобы разобраться в природе того или иного нервно-мышечного заболевания, исследователи стремятся связать их с нарушением функциональности определенных белков. Необходимо более точное понимание взаимодействий и функций белков, вовлеченных в патогенез той или иной мышечной дистрофии. Невозможно переоценить значимость данной информации в разработке терапевтических процедур и инновационных методов лечения большого количества нервно-мышечных заболеваний. Цель проекта В Help Cure Muscular Dystrophy прибегают к помощи World Community Grid в определении белок-белковых взаимодействий более 2 200 безызбыточных белков с известной структурой, информация о которых содержится в Белковом Банке данных (www.rcsb.org/pdb). Исследоваться будут также белки, которые синтезируются в результате экспрессии мутировавших генов. Целью проекта является создание новой базы данных с информацией о функционально взаимодействующих белках. Дальнейшие исследования будут связаны с изучением участков белков, вовлеченных во взаимодействия лигандов (например, лекарств) с ДНК. Эта тема представляет значительный медицинский интерес, хотя сейчас упор делается на проектировании малых молекул (лигандов), которые нарушают (ингибируют) или улучшают работу определенных белков, намного сложнее определить, как та же самая малая молекула может прямо или косвенно влиять на другие взаимодействия белков. Исследовательский подход В этом проекте используется подход, в котором скомбинирована информация об эволюции (как развитие изменило белки и определило их функции) и молекулярное моделирование (вычислительное определение относительного положения двух взаимодействующих белковых партнеров), чтобы идентифицировать потенциальные взаимодействия. Молекулярное моделирование объединяет теоретические методы и вычислительные способы для моделирования поведения биологических молекул. Эти методы и способы применяются для исследования структуры биологических систем, таких как свернувшиеся белки, или при идентификации белков-лигандов, связывающихся с небольшими химическими системами, крупными биомолекулами и белковыми комплексами. Докинг (стыковка) белок-лиганд - молекулярная техника моделирования, которая прогнозирует положение и ориентацию (трехмерную структуру) белка относительно лиганда (который может быть другим белком, ДНК или лекарственным средством). Методы молекулярного докинга основаны исключительно на физических принципах — даже белки с неизвестными или слабо изученными функциями могут быть исследованы на предмет стыковки с различными лигандами. Единственным обязательным условием является знание трехмерной структуры белков, полученное теоретическими методами или в ходе экспериментального исследования. При использовании молекулярного докинга производится перебор известных молекул из баз данных с целью поиска вариантов, обнаруживающих сродство, т.е. хорошо связывающихся друг с другом. Степень сродства определяется с помощью оценочной функции по геометрическим и химическим параметрам полученного соединения. Геометрия оценивается по тому, насколько хорошо сочетаются трехмерные структуры белка и лиганда (должны быть подогнаны как рука под перчатку). С химической точки зрения оценивается сила межатомных взаимодействий между белком и лигандом. Для таких сложных структур как белки (наименьшие из которых содержат сотни атомов) исследования белок-белковых взаимодействий могут потребовать значительных вычислительных ресурсов. Без World Community Grid вычисления, необходимые для моделирования молекулярного докинга, были бы слишком трудоёмкими. Для первых 168 белков, изучавшихся еще в первой фазе проекта, привлеченное с помощью WCG процессорное время составило 8 000 лет. Во второй фазе проекта, где будут исследоваться уже 2 246 белков, предполагаемое время расчетов достигнет 91 680 лет. Преодолеть этот вычислительный барьер поможет информация об эволюции белков, позволяющая предсказывать взаимодействующие участки белков. Этот предварительный анализ уменьшает предполагаемое время вычислений в 100 раз, тем самым увеличивая количество исследуемых белков. Тем не менее, без помощи World Community Grid запланированные вычисления окажутся невыполнимыми. Добровольцы, жертвующие свободные вычислительные ресурсы World Community Grid, высвободят мощности для других исследований, проводимых AFM, CNRS, INSERM и пр. научными игроками, направленных на развитие научных инструментов, которые увеличивают наши знания о природе и методах лечения редких заболеваний. Исследователи Вычислительная биология: * Dr. Alessandra Carbone, Analytical Genomics, FRE3214 CNRS-UPMC, Universitй Pierre et Marie Curie, Paris. * Yann Ponty, Department of Computer Science, Universitй Pierre et Marie Curie, Paris. ГРИД-технологии: * Jean-Marie Chesneaux team, Computer Science Laboratory of Paris 6 (LIP6), CNRS Laboratory UMR 7606, Universitй Pierre et Marie Curie, Paris. Миопатия (заболевание мышц): * Pascale Guicheney team, INSERM Laboratory U582, Myology Institut, "Pitiй-Salpetriиre" Hospital, Paris. Молекулярное моделирование: * Richard Lavery, Institut de Biologie et Chimie des Protйines, CNRS UMR 5086, Universitй de Lyon. * Sophie Sacquin-Mora, Laboratory of theoretical biochemistry, CNRS Laboratory UPR9080, "Institut de Biologie Physico-Chimique", Paris. Вопросы и ответы по проекту: (Show/Hide) Скриншот графического клиента  График производительности проекта:  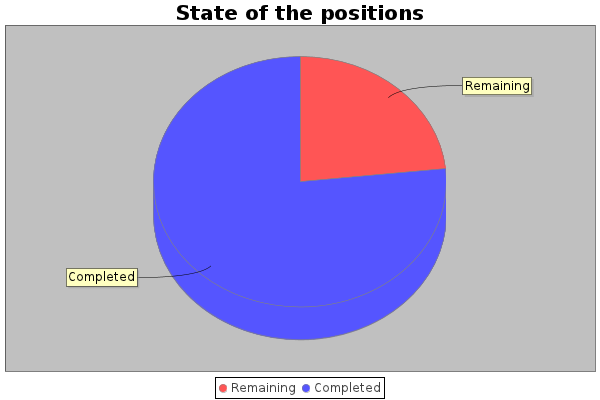 Це повідомлення відредагував Rilian: Dec 11 2010, 14:10 |
   |

 
|



|
  |
Відповідей(30 - 44)
| Rilian |
Feb 4 2011, 19:19
Пост
#31
|
|
interstellar Група: Team member Повідомлень: 17 377 З нами з: 22-February 06 З: Торонто Користувач №: 184 Стать: НеСкажу Free-DC_CPID Парк машин: ноут и кусок сервера |
QUOTE(KING100N @ Feb 4 2011, 14:13)  Обновление статистики - посчитано 56.83% с этой инфой, Sekerob опубликовал рассчет времени кранчинга проекта QUOTE My abacus makes it late Q2-2012, Spring Break and Summer Holiday effects inclusive. This indicator has not moved for quite a few months, hard entered May 31, and computed May 29, subject to change without notice of course  ) )http://www.worldcommunitygrid.org/forums/w...ad_thread,30815 Примерно проект завершится в конце мая 2012 -------------------- |
|
|
|



| corsar83 |
Feb 25 2011, 14:36
Пост
#32
|
 кранчер зі стажем Група: Trusted Members Повідомлень: 426 З нами з: 28-January 10 Користувач №: 1 282 Стать: Чол Free-DC_CPID Парк машин: Pentium E6500 2,93Ghz, Phenom X6 1055T 2.8Ghz Gigabyte 5870OC(875/4800), Radeon 5750 |
Обновление статистики за 18 февраля - 59.78%
 --------------------   |
|
|
|
| Rilian |
Mar 7 2011, 15:34
Пост
#33
|
|
interstellar Група: Team member Повідомлень: 17 377 З нами з: 22-February 06 З: Торонто Користувач №: 184 Стать: НеСкажу Free-DC_CPID Парк машин: ноут и кусок сервера |
Statistics updated on 2011 March 4th : 62.43%
 > Statistics updated on 2011 February 18th : 59.78% -------------------- |
|
|
|
| corsar83 |
Jun 28 2011, 12:29
Пост
#34
|
|
кранчер зі стажем Група: Trusted Members Повідомлень: 426 З нами з: 28-January 10 Користувач №: 1 282 Стать: Чол Free-DC_CPID Парк машин: Pentium E6500 2,93Ghz, Phenom X6 1055T 2.8Ghz Gigabyte 5870OC(875/4800), Radeon 5750 |
Statistics updated on 2011 June 27th : 76.67%
-------------------- |
|
|
|
| Rilian |
Jan 3 2012, 22:37
Пост
#35
|
|
interstellar Група: Team member Повідомлень: 17 377 З нами з: 22-February 06 З: Торонто Користувач №: 184 Стать: НеСкажу Free-DC_CPID Парк машин: ноут и кусок сервера |
July 31, 2010
Hi to all! Thought to try to explain what we are doing right now before you take some vacation, like the scientists here. Hope it will help to feel that things are improving and that the project is very active from this side!! Actually, someone new will join the group on september, Anne Lopes. Anne is assistant professor in structural bioinformatics and has a background in physical-chemistry. She is very interested in working on the protein partnership problem with the numerical approach we developed and on the data analysis of the huge amount of information you are producing! The state of the art here is the following. In the paper [S. Sacquin-Mora, A. Carbone and R. Lavery (2008), Identification of protein interaction partners and protein-protein interaction sites, J. Mol. Biol. 382, p1276-1289] we developed a numerical method to detect protein partners. The method was presented and tested on a small quantity of known protein complexes. As you can imagine, as soon as the data from HCMD Phase 1 arrived (THANKS TO YOUR CONTRIBUTION!!) we retested the approach to verify whether we could confirm the results on a larger dataset. This is indeed the case, the method works, and we can distinguish protein partners within the about 150 proteins tested. We observed that the signal is much less sharp when we work with 150 proteins than with 12 proteins (like in the paper) though and that some extra work should be done to improve the numerical method. Remember that for HCMD Phase 2 we shall search for partners among about 2200 proteins. At the moment, we have improved the formula introduced in the paper and we are developing an "intelligent" approach to arrive fast and surely to identify a small number of potential partners for any protein. Let me give you an insight on the complexity underlying the problem. It has something to do with the understanding of protein population. This is an important point to assimilate, if you like to understand a bit more of our analysis. When we consider a protein, we do not just study one protein (that is, its geometry and its physico-chemical properties: this is already taken into account in the docking algorithm running on your computers and into JET, the program that allowed us to predict protein binding sites) but we rather study its behaviour with the population of proteins that are around it (in the cell; for the HCMD phase 2, population means the 2200 proteins analyzed in your computers). In other words, when we look at a protein we hope to get a signal on its partnership by looking at her way to interact with all other proteins in the population. This means that we hope to learn from bad interactions as well as from good interactions. The information that YOU are giving us provides to us some insight on what is bad and what is good! but this is not enough and we shall use also some extra observation on the interaction of the protein within a population. Some proteins are slippery, meaning that they do not seem to glue to any partner. Some others are gluing, meaning that they do glue to essentially everybody. Then there are many other proteins (about a half) that seem to stick on the right place with some specificity. They are the easiest to study. When we use, in our calculations, contributions coming from the entire population, one should think that these contributions come, in principle, from slippery proteins, gluing proteins and many other proteins whose behaviour is less sharply characterizable. "Noise" might enter into the calculation and we wish to reduce it. Learning from the whole set of interactions of a protein, means to learn to which group the protein belongs to. Once this is determined, the numerical criteria that we developed could be adjusted to accurately predict a partner or a small set of potential partners, whenever possible. The understanding of the whole set of behaviours that we need to take into account to know how to correctly evaluate the data coming from WCG is our goal today. There are a few other concerns that are present in our analysis, and they have something to do with : 1. the algorithmic aspects concerning the handling of large amount of information to be combined for the "learning" approach I mentioned above. 2. the fact that on HCMD phase 2 data analysis, we use JET predictions of protein interaction in our numerical criteria instead of actual real interfaces as done in the paper cited above. This implies a loss of precision that we should consider in our numerical evaluations of the interactions. These informations should give you some insight on the complexity of the question we face today. Hope that everyone will be feeling that we are advancing, together, for a project that runs alive and hopefully will reserve exciting surprises to all. We expect it. Have a good summer! Alessandra -------------------- |
|
|
|
| Rilian |
Jan 3 2012, 22:56
Пост
#36
|
|
interstellar Група: Team member Повідомлень: 17 377 З нами з: 22-February 06 З: Торонто Користувач №: 184 Стать: НеСкажу Free-DC_CPID Парк машин: ноут и кусок сервера |
UPDATES for the project
December 21, 2011 Hi to all, with Sophie and Richard we have written up an account of the docking analysis of the 168 protein complexes of the Mintseris dataset tested in phase 1. The paper is under review right now and will give you the link as soon as it is published. The analysis of the dataset of 168 protein complexes is not finished yet! In fact, we try to improve the signals for the detection of partnership. There are two main points that one needs to keep in mind. In phase 2 we do not know the real partners and we had to use predictions of interaction sites to run MaXDO. This was because the search space on a protein surface could not be exhaustively explored, even with the help of WCG. it would be far too big! This means that we need to understant on a pool of proteins that we know (that is the 168 protein complexes) how the predictions of protein interaction sites will impact partners predictions. This is what we carefully investigate right now. It takes time! There are a number of intermediate results that you might like to know about: 1. the analysis realized in [Sacquin-Mora et al. 2008] on 12 complexes, has been scaled to 168 complexes, and it highlighted a predictive protein-protein interaction power of AUC=0.84 (see Figure A below) when using knowledge on real interaction surfaces and when exploring the whole protein surface. It is important to stress that this successful scaling of the analysis in [Sacquin-Mora et al. 2008] to 168 proteins was not an obvious guess! Why successful? The AUC is a probability measure used to evaluate the accuracy of the test. Values vary from 0 to 1, where 1 represents a perfect test and 0.5 represents a worthless test. Roughly speaking, one can think of the following ranking: .90-1 = excellent, .80-.90 = good, .70-.80 = fair, .60-.70 = poor, .50-.60 = fail. 2. We also observed that amongst the 168 protein complexes several had the tendency to bind to nearly all other proteins and others showed very few strong interactions. Both these families of proteins negatively contribute to partnership prediction, and, when eliminated, enable the predictive power to be increased to an AUC=0.98 (see Figure B). 3. When experimental information on interaction surfaces is replaced with data from JET [Engelen 2009] (the tool for conservation analysis developed within our consortium) the predictive power only decreases slightly, with an AUC=0.82. This suggests that coupling protein interface predictions with docking is a very promising approach. 4. Nevertheless improvements are still required, since when JET predictions are used to delimit the docking area, as well as to compute the numerical index that discriminates partners, the predictive power falls at an AUC=0.59. This implies that better interaction patch detection has to be developed. However, we note that a subgroup of 20 complexes was identified where JET predictions already yielded very good predictions (AUC=0.97; Figure C below), suggesting that generating subgroups by categorizing protein interaction proclivities could improve performance. 5. Lastly, we systematically analyzed complexes in terms of the functional classes of the interacting proteins. The complexes could be grouped into: Enzyme-Inhibitors (46 proteins), Antigen-Antibody (20), Antigen-Antibody Bound (24), Others (78), and also as, Rigid Body (126), Medium (26) and Difficult (16). Interactions within certain classes, such as Enzyme-Inhibitors, were clearly easier to predict suggesting that such classifications should be considered in partnership prediction. 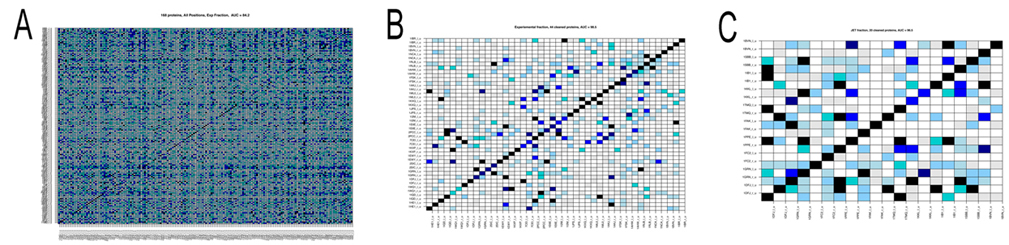 Figure. Matrices of pairwise interaction indexes for different subsets of proteins. High interaction scores (between 0.7 and 1, blue and black in the color scale) indicate a high probability of interaction. Proteins are ordered in the matrix such that true interacting partners lie on the diagonal. A: full dataset of 168 protein complexes. Interaction scores were computed using knowledge of the experimental interfaces (AUC=0.84). B: subset of 44 protein complexes leading to an AUC=0.98. Interaction scores were computed using knowledge on experimental interfaces. C: subset of 20 proteine complexes leading to an AUC=0.97. Interaction scores were computed using interfaces predicted by JET. At the moment we work on coevolution between protein interfaces and on improving JET interaction predictions. With both advancements we count improving identification of new partners, and increase the AUC above. We have done a lot of work already on this. A new approach to coevolution analysis, treating especially conserved sites like protein interfaces, has been recently developed at the lab. More on this soon. Merry Christmas and a Happy New Year to all! Alessandra -------------------- |
|
|
|
| KING100N |
Jan 27 2012, 18:09
Пост
#37
|
 кранчер з фермою Група: Trusted Members Повідомлень: 788 З нами з: 17-October 08 Користувач №: 847 Стать: Чол |
До конца проекта осталось 18 дней:
QUOTE Hello all, This is the official statement saying there is an estimated 18 days left of new work to be sent out. In addition, we will be changing the way work units are distributed for this project. Previously priority was given to work units that were children/grandchilden of the original parent work units. We will be changing this to give priority to parent work units (so these will be sent prior to children/grandchild work units). This will allow for a more consistent supply of hcmd2 work units until the end of the project. Thank you for all your help and support with this project! Seippel https://secure.worldcommunitygrid.org/forum...ad,32499#361380 --------------------  |
|
|
|
| corsar83 |
Jan 27 2012, 18:18
Пост
#38
|
|
кранчер зі стажем Група: Trusted Members Повідомлень: 426 З нами з: 28-January 10 Користувач №: 1 282 Стать: Чол Free-DC_CPID Парк машин: Pentium E6500 2,93Ghz, Phenom X6 1055T 2.8Ghz Gigabyte 5870OC(875/4800), Radeon 5750 |
Отличная новость. Жаль не досчитаю до сапфира, 3 месяца расчётов еще нужно
 UPD. Или ще работ подвалят чото не понял -------------------- |
|
|
|
| KING100N |
Jan 27 2012, 18:33
Пост
#39
|
|
кранчер з фермою Група: Trusted Members Повідомлень: 788 З нами з: 17-October 08 Користувач №: 847 Стать: Чол |
QUOTE(corsar83 @ Jan 27 2012, 18:18) Или ще работ подвалят чото не понял Да вроде нет, хотя, может и насыпят охапку, как в HFCC -------------------- |
|
|
|
| Rilian |
Jan 27 2012, 23:01
Пост
#40
|
|
interstellar Група: Team member Повідомлень: 17 377 З нами з: 22-February 06 З: Торонто Користувач №: 184 Стать: НеСкажу Free-DC_CPID Парк машин: ноут и кусок сервера |
У меня сапфир есть - любезно кранчу другие проекты, чтобы предоставить шанс охотникам за бейджи
 -------------------- |
|
|
|
| Rilian |
Jan 30 2012, 11:53
Пост
#41
|
|
interstellar Група: Team member Повідомлень: 17 377 З нами з: 22-February 06 З: Торонто Користувач №: 184 Стать: НеСкажу Free-DC_CPID Парк машин: ноут и кусок сервера |
Спешите докранчить до бейджика (минимум 14 процессорных дней)! Больше такой возможности в жизни не будет!
-------------------- |
|
|
|
| corsar83 |
Jan 30 2012, 12:02
Пост
#42
|
|
кранчер зі стажем Група: Trusted Members Повідомлень: 426 З нами з: 28-January 10 Користувач №: 1 282 Стать: Чол Free-DC_CPID Парк машин: Pentium E6500 2,93Ghz, Phenom X6 1055T 2.8Ghz Gigabyte 5870OC(875/4800), Radeon 5750 |
Уже поздно
 . Я не успею, эхх.. . Я не успею, эхх..-------------------- |
|
|
|
| Rilian |
Jan 30 2012, 12:31
Пост
#43
|
|
interstellar Група: Team member Повідомлень: 17 377 З нами з: 22-February 06 З: Торонто Користувач №: 184 Стать: НеСкажу Free-DC_CPID Парк машин: ноут и кусок сервера |
corsar83, Еще время есть. Подтягивайся
-------------------- |
|
|
|
| corsar83 |
Jan 30 2012, 12:34
Пост
#44
|
|
кранчер зі стажем Група: Trusted Members Повідомлень: 426 З нами з: 28-January 10 Користувач №: 1 282 Стать: Чол Free-DC_CPID Парк машин: Pentium E6500 2,93Ghz, Phenom X6 1055T 2.8Ghz Gigabyte 5870OC(875/4800), Radeon 5750 |
Мне нужно еще 80 дней расчетов, вроде и не много, но.... Круглосуточно считать не могу, так может и успел бы
 -------------------- |
|
|
|
| KING100N |
Jan 30 2012, 13:26
Пост
#45
|
|
кранчер з фермою Група: Trusted Members Повідомлень: 788 З нами з: 17-October 08 Користувач №: 847 Стать: Чол |
corsar83,
Давай слабый ключ от учетки. Со вторника, надеюсь, я снова в строю, помогу 8ю ядрами -------------------- |
|
|
|
|
|
|
1 Користувачів переглядають дану тему (1 Гостей і 0 Прихованих Користувачів)
0 Користувачів:

|
Lo-Fi Версія | Поточний час: 16th April 2024 - 16:24 |