
Реферат
«Моделювання тривимірної структури білків у проекті Rosetta@Home»
Вступ
Проект проводиться лабораторією доктора Девіда Бейкера, яка є частиною факультету біохімії Університета Вашингтона. Ціль проекту Rosetta@Home – розробити покращену модель внутрішньо та міжмолекулярних взаємодій, та використати цю модель для передбачення та проектування макромолекулярних структур та взаємодій.Розроблені алгоритми для проектування та передбачення структур можуть уявляти собою великий біологічний зміст. Вони забеспечують строгі та обєктивні тести, які покращують модель та збільшують фундаментальне розуміння.
Проект використовує комп'ютерну програму на ім'я Розетта, щоб виконувати обчислення білка і дизайну. У ядрі Розетти є потенційні функції для обчислення енергії взаємодій в межах і між макромолекулами, і методи для пошуку структури з найнижчою енергією для послідовності амінокислот (прогноз структури білка) або комплексу білок-білок, і для пошуку послідовності амінокислот з найнижчою енергією для білка або комплексу білок-білок (проектування білка). Зворотний зв'язок від тестів прогнозу і проектування використовується безперервно, щоб поліпшити потенційні функції і алгоритми пошуку. Розвиток однієї комп'ютерної програми для обробки цих різноманітних проблем має значні переваги: по-перше, різні додатки забезпечують додаткові тести основної фізичної моделі (фундаментальна фізика / фізична хімія, звичайно, одна і та ж у всіх випадках); по-друге, багато проблем, що представляють поточний інтерес, типа проектування гнучкого базового білка і стиковки білок-білок з базовою гнучкістю, залучають комбінацію різних методів оптимізації.
1. Проектування структури білків
Протягом останніх декількох років проект використовує обчислювальний метод проектування білка, щоб істотно стабілізувати декілька маленьких білків, перепроєктіруя кожен залишок їх послідовностей, щоб перепроектувати пристрій основи білка, перетворити мономірний білок в «strand-swapped dimer», і термостабілізіровать фермент. Основним моментом була модернізація шляху згортання білка G, маленького білка, що містить дві бета-шпільки, відокремлені альфа-спиралью. У білці природного походження, зруйнована перша шпилька, а друга шпилька сформована на швидкості, що обмежує крок в згортанні. У перепроектованому варіанті, в якому значно стабілізована перша шпилька і дестабілізується друга шпилька, порядок подій повністю змінений: перша шпилька сформована, а друга шпилька зруйнована в стані переходу, що згортається. Здатність перепроектувати шляхи, що згортають білок, раціонально показує, що наше розуміння детермінантів згортання білка значно просунулося.

Мал.2.1 Проект білків і белок-белкових взаємодій з високою розподільною здібністю. Порівняння проектної моделі і кристалічної структури (зліва) інтерфейсу нової розробленої ендонуклеази з новою специфікою розколу ДНК, і (справа) знов розроблений білок TOP7.
Що особливо захоплює з недавніх пір - створення нових білків з довільно вибраними тривимірними структурами. Ми розвивали загальну обчислювальну стратегію для того, щоб створити ці структури білка, яка включає повну гнучкість основи в оптимізацію послідовності, що ротамер-базується. Це було досягнуто об'єднанням в Розеттє прогнозу структури білка методом ab initio, обробки енергії атомного рівня, і проектування послідовності. Процедура використовувалася для проектування білка з 93 залишками на ім'я TOP7 з новою послідовністю і топологією. TOP7, як визначили, був мономірним і згорнутий, і рентгенокрісталична структура TOP7 дивно схожа (RMSD = 1.2; див. праву групу малюнка 2.1) на проектну модель. Проект нового кулястого згортання білка і близька відповідність кристалічної структури з проектною моделлю має широкі значення для проектування білка і прогнозу структури білка і відкриває двері в дослідження великих областей всесвіту білка, ще не спостережуваних в природі.
2. Проектування протеїн-протеїнових взаємодій
Щоб розширити ці методи на белок-белковиє взаємодії і особливо на модернізацію специфіки взаємодії, вибрано високо-подібний комплекс з colicin E7 дезоксирібонуклеаза і її спорідненого інгибіторного імунного білка як систему-зразок. Використовано фізичну модель, описану вище і модифікацію нашої обчислювальної проектної стратегії на основі пошуку ротамеров, щоб провести нові пари білка інгібітору дезоксирібонуклеази, передбачених, щоб сильно взаємодіяти один з одним, але не з білками дикого типу. Розроблені білкові комплекси мають субнаномолярну схожість, функціональну і визначену в природних умовах, і мають більше на порядок у відмінності властивостей між спорідненими і неспорідненими парами в штучних умовах. Цей підхід повинен бути застосовний до проектування взаємодіючих пар білка з новими специфіками для того, щоб описати і відтворити мережі взаємодії білка в живих клітинах.
У співпраці з дослідницькими групами Баррі Стоддарда (Barry Stoddard) і Рея Монната (Ray Monnat) (Дослідницький центр Раку Фреда Хачинсона - Fred Hutchinson Cancer Research Center), проведено штучну, високоспецифічну ендонуклеазу, плавлячи області ендонуклеаз I-DmoI і I-CreI, що повертаються, за допомогою обчислювальної оптимізації нового інтерфейсу домен-домен між їх зазвичай невзаємодіючими білками. Фермент, що вийшов, E-DreI (Engineered I-DmoI/I-CreI), пов'язує довгу химерну цільову ділянку ДНК з наномолярними властивостями, розколюючи його точно по нормі, еквівалентній його природним батькам. В даний час розробники проекту пробують провести нові ендонуклеази, розширюючи методологію проектування на інтерфейси белок-нуклєїнова кислота, щоб перепроектувати інтерфейс білок-ДНК. У обох з цих систем було можливо визначити рентгенокрісталічні структури розроблених комплексів. Як у разі TOP7, фактичні структури виявилися дуже схожими на спроектовані моделі (малюнок 2.1, ліва група), що затверджує точність нашого підходу моделювання з високою роздільною здатністю.
3. Передбачення тривимірної структури білка
Картина згортання білка, яка мотивує підхід в прогнозі третинної структури білка ab initio, - це те, що послідовно-залежні місцеві взаємодії зміщують сегменти ланцюжка до типових відмінних наборів місцевих структур, і що нелокальні взаємодії вибирають найнижчі вільно-енергетичні третинні структури з багатьох формацій, сумісних з цими місцевими ухилами. У здійсненні стратегії, запропонованої цією картиною, використовують різні моделі, щоб розглянути локальні і нелокальні взаємодії. Замість того, щоб застосовувати фізичну модель для локальних відносин структура-послідовність, звертаються до бази даних білків і беремо розподіл локальних структур, прийнятих короткими сегментами послідовностей (завдовжки менше 10 залишків) у відомих тривимірних структурах як наближення до розподілу структур, вибраних ізольованими пептидами з відповідними послідовностями. Дані первинні нелокальні взаємодії - hydrophobic burial, електростатика, водневий зв'язок головного ланцюга, і виключений об'єм. Структури, які є одночасно сумісними і з локальними ухилами структури послідовності, і з нелокальними взаємодіями, проведені при використанні модельованого відпалу, щоб мінімізувати нелокальну енергію взаємодії в місці, визначеному локальними структурними розподілами.
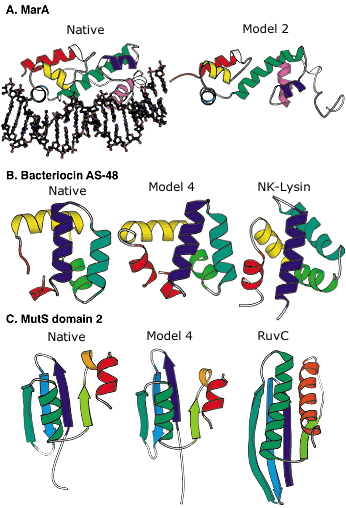
Малюнок 4.1. Сліпі прогнози структури білка від CASP3 і CASP4.
A: Зліва кристалічна структура чинника транскрипції MARA, пов'язаного з ДНК; справа краща запропонована проектом модель в CASP3. Не дивлячись на багато неправильних деталей, повне згортання передбачене з достатньою точністю, щоб дозволити зрозуміти суть способу скріплення ДНК.
B: Зліва кристалічна структура bacteriocin AS-48; в середині краща запропонована проектом модель в CASP4; справа структурно і функціонально зв'язаний білок (NK-lysin), ідентифікований з використанням цієї моделі в пошуку на основі структури в Банці даних Білка (PDB). Структурна і функціональна подібність не розпізнаються з використанням методів порівняння послідовності (ідентичність між двома послідовностями - тільки 5 відсотків).
C: Зліва кристалічна структура другої області MUTS; в середині краща запропонована проектом модель для цієї області в CASP4; справа структурно зв'язаний білок (RUVC) із зв'язаною функцією, розпізнаною з використанням моделі в пошуку на основі структури PDB. Подібність не була розпізнана з використанням порівняння послідовності або методів розпізнавання згортання.
Розетта була перевірена в дворічних експериментах CASP (критична оцінка прогнозу структури), в яких провісникам кидають виклик зробити сліпі прогнози структур прийнятими послідовностями білка, структури яких були визначені, але ще не видані. Починаючи з CASP3 в 1998, Розетта послідовно була головним методом виконання для прогнозу ab initio, як повідомлено незалежними експертами. У експерименті CASP4, наприклад, Розетта була перевірена на 21 білці. Прогнози для цих білків, які відчувають нестачу у виявленій подібності послідовності будь-якому білку із заздалегідь певною структурою, мали безпрецедентну точність і послідовність. (Деякі приклади показані на малюнку 4.1.) Чудові прогнози були також зроблені в експериментах CASP5 і CASP6. Заохочені цими багатообіцяючими результатами, працівники проекту провели моделі для всіх великих сімей білка завдовжки менше 150 амінокислот.
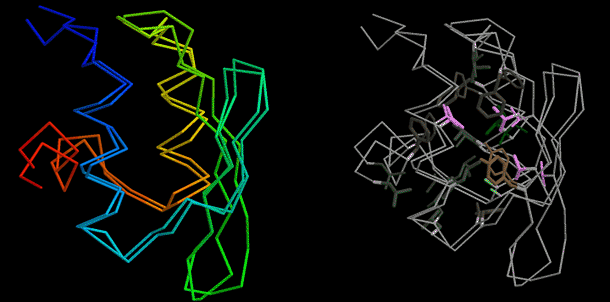
Малюнок 4.2. перший, близький до розміру атомного рівня, сліпий ab initio прогноз структури - CASP6 T281. Методологія обробки з високою розподільною здатністю, описана в тексті, створила зразковий 1.5-A RMSD від кристалічної структури (ліва група), з аспектами природної упаковки бічного ланцюга (права група).
Основним моментом CASP6 був перший de novo сліпий прогноз, який використовував методологію обробки з високою роздільною здатністю, щоб близько досягти точності з високою роздільною здатністю. Відносно коротка послідовність (76 залишків) дозволила застосовувати методологію все-атомної обробки не тільки до природної послідовності, але також і до послідовності багатьох гомологов. Центр групи структур з найнижчою енергією, як виявилось, був дивно близький до природної структури (1.5 A; малюнок 4.2). Протокол обробки з високим дозволом зменшив RMSD з 2.2 A до 1.5 A, а бічні ланцюги упаковуються в ядрі білка в манері, частково подібній природній (малюнок 4.2, права група).
Було розширено стратегію ab initio Розетти для прогнозу структури на проблему використання обмежених експериментальних даних, щоб провести моделі білків. Включаючи хімічний зсув і інформацію NOE і пізнішу інформацію двополюсного зчеплення в процедуру генерації структури Розетти, працівники проекту були в змозі провести набагато точніші моделі, ніж з прогнозом однієї структури ab initio або використовуючи ті ж самі обмежені набори даних з методології генерації структури звичайним ядерним магнітним резонансом (ЯМР). Захоплююча недавня розробка полягає в тому, що процедура Розетти може також використовувати в своїх інтересах непризначені дані ЯМР і отже обійти важкий і довгий крок призначення спектрів ЯМР.
Метод ab initio структурного прогнозу Розетти, ЯМР метод визначення структури на базі Розетти, і новий метод для порівняльного моделювання, які використовують de novo підхід Розетти для моделювання частини структури (перш за все довгі петлі), які не можуть бути змодельовані точно, засновані на шаблоні структури гомолога, були всі здійснені в суспільному сервері на ім'я Robetta. Цей сервер, який має постійне відставання користувачів у всьому світі, був одним з кращих серед всіх повністю автоматизованих серверів прогнозу структури в тестах CASP6 і CASP5.
4. Передбачення білок-білкових взаємодій
Протягом безлічі років ми впливали на обробку структури білка - стимулююча проблема через велику кількість мір свободи. Автори проекту зацікавилися белок-белковою стиковкою тому, що два білкові партнери, приблизно, не піддаються істотним конформаційним змінам протягом стиковки, місце, яке буде обстежено, - шість мір свободи твердого тіла на додаток до мір свободи бічного ланцюга - є набагато меншим. Тоді як це саме по собі важливо, ця проблема - хороший твердий крок до складнішої проблеми обробки структури.
Було розроблено новий метод, щоб передбачити белок-белковиє комплекси з координат незв'язаних мономірних компонентів. Цей метод реалізує твердотільний, з низьким дозволом, пошук Монте Карло, що супроводжується одночасною оптимізацією зсуву основи і пристроїв бічного ланцюга з процедурою мінімізації Монте Карло і фізичною моделлю, використовуваною в нашій роботі прогнозу структури з високою роздільною здатністю. Одночасна оптимізація мір свободи бічного ланцюга і твердого тіла контрастує з більшістю інших поточних підходів, які моделюють белок-белковую стиковку як проблему відповідності форми твердого тіла, з бічними ланцюгами, встановленими, що зберігаються. Розробники проекту недавно поліпшили метод (RosettaDock), розробивши алгоритм, який дозволяє ефективне здійснення вибірки не-ротамер пристроїв бічного ланцюга під час стиковки.
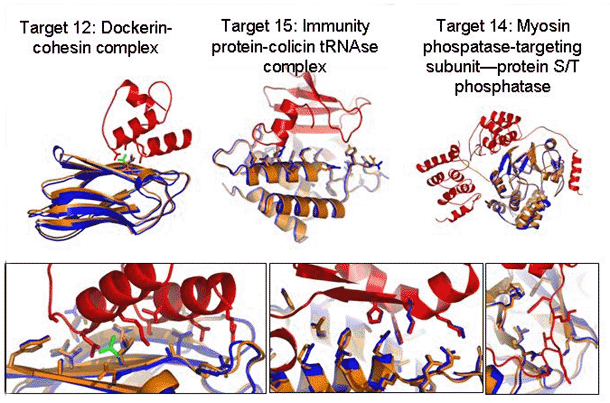
Малюнок 5.1. Результати белок-белкової стиковки КАПРІ (критична оцінка передбачених взаємодій). Суперпозиція структур передбаченого (синій) і рентгенівського (червоний і оранжевий) білкового комплексу. Зелений - бічний ланцюг, пристрій якого був правильно передбачений, щоб змінитися після складного формування. Верхня група, цілий комплекс. Нижня група, деталі інтерфейсу. На додаток до орієнтації твердого тіла, пристрої більшості бічних ланцюгів передбачені правильно.
Сила RosettaDock була висунута на перший план в недавньому змаганні КАПРІ по сліпій белок-белкової стиковці, яке було проведено в грудні 2004г. У КАПРІ провісникам дають структури двох білків, відомих, що вони формують комплекс, і пропонують передбачити структуру комплексу. Прогнози RosettaDock для цілей без істотних конформаційних змін підстави були вражаючі (як показано на малюнку 5.1). Були не тільки орієнтації твердого тіла двох партнерів, передбачених майже абсолютно, але також і майже всі бічні ланцюги інтерфейсу були змодельовані дуже точно. Ці правильні моделі ясно виділилися енергетично нижче, ніж всі інші моделі, які ми провели, що припускає, що потенційна функція достатньо точна.
Ці багатообіцяючі результати припускають, що даний метод може скоро стати корисним для того, щоб провести моделі біологічно важливих комплексів із структур ізольованих компонентів, і ширше припускають, що моделювання з високою роздільною здатністю структур і взаємодій є в межах досяжності. Ясна мета для роботи прогнозу Rosetta для мономірних структур полягає в тому, щоб досягти рівня точності цих моделей.
5. Покращення фізичної моделі
Поточний підхід проекту в поліпшенні енергетичних функцій залучає комбінацію квантових хімічних обчислень на простих зразкових складах, традиційних підходах молекулярної механіки, і аналізу білкових структур. Розробники проекту використовували такий підхід, щоб розробити покращуваний потенціал водневого зв'язку. Особливо відомий результат полягає в тому, що залежність орієнтації водневого зв'язку в квантових хімічних обчисленнях на формамідних дімерах є дивно схожою на відмічену у водневих зв'язках бічний ланцюг - бічний ланцюг в структурах білка, але відмінною від цього в силових полях в поточній молекулярній механіці, яка нехтує ковалентним характером водневого зв'язку. Зворотний зв'язок від обчислень прогнозу і проектування забезпечив безперервний стимул і керівництво для поліпшення енергетичної функції; наприклад, невідповідності в нашій обробці белок-белкових взаємодій привели до недавнього розвитку моделі, що ротамер-базувалася, для встановлених водою водневих зв'язків.
6. Плани на майбутнє
Методи прогнозу і проектування тепер досягли пункту, де вони можуть бути застосовані до важливих біологічних проблем. Особливо обнадіює після років робіт над моделюванням з високим дозволом близькість до прогнозів з атомарним дозволом структур комплексів в КАПРІ (малюнок 5.1), знов прогноз 1.5-A в CASP6 (малюнок 4.2), і близька згода для TOP7 (малюнок 2.1, справа) і проектних моделей белок-белкового інтерфейсу (малюнок 2.1, зліва) з рентгенокрісталічними структурами. Ці результати говорять про те, що моделювання з високою точністю починає працювати.
За наступні декілька років, автори проекту прагнуть покращувати і розширювати методи прогнозування. Розробники особливо зосереджені на поліпшенні точності прогнозу структури з високою роздільною здатністю (яка потрібна, якщо моделі повинні бути взагалі корисними). Щоб досягти цього, вони працюють, щоб поліпшити основну фізичну модель і методологію здійснення вибірки. Автори також розвивають покращувані методи, щоб передбачити і перепроектувати специфіку взаємодії БІЛОК-ДНК, і розширюють методологію проектування білка на проектування ферментів, які каталізують хімічні реакції, що не каталізують білками природного походження.
7. Дослідження, пов'язані з хворобами
Коментарі Девіда Бейкера, професора біохімії Університету Вашингтона (the University of Washington)
Моя дослідницька група займається і дослідженнями фундаментальних методів розвитку, і більш безпосередньо намагається боротися з хворобами. Більшість інформації на цьому сайті сосредотачиваєтся на фундаментальних дослідженнях, але я думав, що Вам може бути цікаво почути про ту частину нашої роботи, пов'язаної з лікуванням хвороб, до якої Ви вносите свій внесок, беручи участь в проекті Rosetta@home.
Рак (Cancer)
Рак може бути викликаний мутаціями в ключових генах, які руйнують нормальні клітинні процеси управління. Ми розробляємо методи для того, щоб вирізувати ДНК на певних ділянках генома, і ми визначатимемо ділянки, які залучені в рак. Після того, як ці ділянки вирізані, вони повинні бути відновлені кліткою, використовуючи другу, незмінену копію гена, і клітка більше не повинна бути злоякісною. Це дуже специфічна форма генотерапії, яка, у разі успіху, обійде основну проблему сучасних методів генотерапії; а саме, сучасні методи вставляють незмінену копію гена безладно в геном, і якщо вставка відбулася біля ракового гена (oncogene), то генотерапія виправить одну хворобу, але викличе іншу. Оскільки наші методи будуть націлені на певні ділянки замість випадкових, вони повинні уникнути цієї пастки.
Рак простати (Prostate Cancer)
Рецептор андрогена (AR) зв'язує тестостерон і відповідає за нормальний чоловічий розвиток. Коли AR стає надчутливим до тестостерону, результатом є рак простати. Сучасне лікування раку простати, зване "гормональна терапія", полягає в пониженні кількості доступного тестостерона (іноді шляхом кастрації). Багато злоякісних пухлин є стійкими до цієї терапії, тому ми застосовуємо нашу методологію моделювання білка так, щоб знайти різні способи відключити AR і вилікувати рак простати. Поза сумнівом, ми пробуємо проектувати білки, які відключать AR навіть у присутності тестостерона. Ми робимо це, проектуючи білки, які перешкоджатимуть проникненню AR в ядро клітки (який проникає туди, щоб зробити свою брудну роботу), і також перешкоджати йому зв'язувати ДНК і активізувати визначені для пухлини гени, навіть якщо він проникне в ядро.
ВІЧ (HIV)
Одній з причин, чому ВІЧ є таким смертельним вірусом, є те, що він розвивався, щоб обдурити відповідну імунну систему. Ми співробітничаємо з дослідниками в Сієтлі і в NIH, щоб спробувати розробити вакцину для ВІЧ. Наша роль в цьому проекті є центральною - ми використовуємо РОЗЕТТУ, щоб проектувати маленькі білки, по вказівці яких імунна система може легко розпізнати невелику кількість критичних областей в оболонці ВІЧ-білка, і згенерувати антитіла. Наша мета полягає в тому, щоб створити маленькі стійкі вакцини білка, які можуть бути достатньо дешеві і проведені у всьому світі.
Малярія (Malaria)
Ми є частиною сумісного проекту, очолюваного Остіном Бертом (Austin Burt) з Лондонського Імперського Коледжу, який є одним з учасників Фонду Гейтса "Основні Проблемні Проекти Глобального Здоров'я (Grand Challenge Projects in Global Health)". Малярія викликається паразитом, який проводить частину свого життєвого циклу в москітові, і вводиться людям комариними укусами. Ідея проекту в тому, щоб зробити москітів, стійкими до паразита, усуваючи гени, потрібні для життя паразита в москітові. Наша частина проекту полягає у використанні базових методів моделювання на наших комп'ютерах (РОЗЕТТА), щоб проектувати нові ферменти, які визначатимуть адресат і деактивувати ці гени.
Сибірська виразка (Anthrax)
Ми використовуємо РОЗЕТТУ, щоб допомогти дослідницькій групі Джона Колліра (John Collier) з Гарварду в моделюванні токсину сибірської виразки, яке повинне внести свій внесок до розробки лікування. Ви можете прочитати резюме, що описує частину цієї роботи.
Хвороба Альцгеймера (Alzheimer's disease)
Причиною Болезні Альцгеймера і багатьох інших хвороб, ймовірно, є білок abberant, який при згортанні формує великі об'єднані структури, названі амілоїд (amyloids) замість того, щоб згортатися в їх нормальні біологічно активні стани. Велике просування було зроблене недавно дослідницькою групою Девіда Ейзенберга (David Eisenberg) в UCLA в рішенні першої структури амілоїда. Ми співробітничаємо з їх дослідницькою групою, щоб використовувати цю структуру для прогнозу того, які частини білків, можливо, формують амілоїди, і це буде першим кроком до блокування амілоїдних формувань і, ми сподіваємося, до лікування хвороби.
Інші віруси
Ми
співробітничали з лабораторією Пема Б'еркмана (Pam Bjorkman) в
Каліфорнійському Технологічному Коледжі (Cal Tech), щоб використовувати
методологію РОЗЕТТИ по стиковці білка з білком, для побудови моделей
білків простого вірусу герпесу в комплексі з людськими білками.
Вищезазначені проекти не виконуються в даний час на BOINC, тому що ми поки що не маємо ефективної системи організації черги, яка дозволяє людям легко поміщати завдання, але сподіваємося найближчим часом добитися цього! Крім того, можете бути упевнені, що обчислення прогнозу структури, що виконуються в даний час на ваших комп'ютерах, матимуть прямий вплив на методи лікування хвороб. Є потрійне пояснення цих прямих взаємозв'язків між прогнозом структури і лікуванням хвороби.
8. Структурний прогноз
Структурний прогноз і конструкція білка тісно зв'язані.
Удосконалення в прогнозі структури приводять до удосконалень в проектуванні білка, який у свою чергу може безпосередньо відтранслювати в створення нових ферментів, вакцин, і т.д. Для отримання додаткової інформації по проектуванню білка Ви могли б звернутися до наукового огляду, який ми недавно написали, і який доступний на нашій домашній сторінці.
Структурний прогноз визначає цілі для нових ліків.
Коли ми передбачаємо структури для білків в людському геномі у великому об'ємі, ми дізнаємося про функції багатьох білків, які допоможуть в розумінні того, як клітки працюють і як відбувається хвороба. Крім того, ми будемо в змозі ідентифікувати багато нових потенційних лікарських цілей, для яких можуть бути спроектовані маленькі молекулярні інгібітори (ліки). Можна сказати, що один з основних шляхів в розробці нових методів лікування людських хвороб, це ідентифікація нових "лікарських" цілей білка. Більшість нових ліків в наші дні взаємодіють з тими ж самими цілями, як і старі препарати, таким чином ці ліки приводять тільки до невеликих удосконалень в лікуванні хвороби. Прогноз структури допомагає нам ідентифікувати нові цілі для препарату, і таким чином допоможе нам знайти інноваційні (можливо навіть зробивши відкриття) методи лікування хвороб.
Структурний прогноз дозволяє використовувати "раціональне проектування" для створення нових ліків.
Якщо ми знаємо структуру білка, ми можемо визначити його функціональні ділянки, і особливо визначити ті ділянки, які потрібно відключити новим препаратом. Обчислення того, чи з'єднається маленька молекула препарату з білковим об'єктом і чи буде він відключений, подібно до різних способів обчислення прогнозу структури, які ми робимо тут, - це в основному проблема виявлення структури з найнижчою енергією білка плюс система препарату - і ми недавно розробили новий модуль в РОЗЕТТІ для вирішення проблеми такого з'єднання. Результати дуже перспективні, і в найближчому майбутньому ваші машини ймовірно виконуватимуть рассчети приєднання ліків, разом з проектами пошуку вакцин і проектування терапевтичного білка, описаних вище, на додаток до обчислень згортання білка, які Ви робите зараз.
9. Короткий словник термінів
Що таке Rosetta?
Rosetta - це програма для прогнозу і проектування структур і комплексів білка - одній з святих чаш Грааля обчислювальної біології.
Що таке білок (протеїн)?
Білок це полімер з амінокислот, закодований геном. Для всіх організмів, що живуть, білки - не тільки будівельні блоки життя, але і мініатюрні молекулярні машини, які виконують майже всі важливі функції хімічного і біологічного взаїмодейстівія в процесі життєдіяльності.
Що таке амінокислоти?
Амінокислоти це хімічні деталі, з яких формуються основні стандартні блоки білків. Завдяки недавно завершеному проекту "Геном людини" відомі амінокислотні послідовності всіх білків в людському організмі. Є 20 різних амінокислот, які визначені генетичним кодом. Ці 20 амінокислот відносяться до різних груп, заснованих на їх хімічних властивостях: кислою або лужною, гідрофільною (що любить воду) або гидрофобной (жирною).
Що роблять білки?
Білки виконують багато основних функцій в клітках живих організмів. Вони копіюють і підтримують геном (ДНК), допомагають кліткам рости і ділитися, заважають їм рости дуже багато, дають клітці її ідентичність (печінка, нейрон, підшлункова залоза, і т.д.), допомагають кліткам спілкуватися один з одним. Білки, коли вони видозмінюються або коли зачеплені токсинами можуть викликати хворобу, типа раку або Альцгеймера. Бактерійні і вірусні білки можуть захопити клітку і знищити її. Коротше кажучи, білки роблять все.
Як білки виконують всі їх різні функції?
Кожен білок згортається в унікальну 3-мірну форму, або структуру. Ця структура визначає функцію білка. Наприклад, білок, який руйнує глюкозу в клітці, здатний використовувати енергію, збережену в цукрі, матиме форму, яка розпізнає глюкозу і зв'язує її (як замокнув і ключ). Він матиме хімічно реактивні амінокислоти, які реагуватимуть з глюкозою і руйнуватимуть її, звільняючи енергію.
Чому білки згортаються в унікальні структури?
Вже давно відомо, що для більшості білків найбільш стійкий стан - в термодинамічному мінімумі. Є на увазі, що унікальна структура білка - найстійкіший стан, який він може прийняти. Представте кулю в трубі - куля завжди котитиметься вниз до підстави труби, тому що це - найстійкіший стан.
Які сили визначають природну унікальність (найстійкішу) структуру білка?
Знання послідовності амінокислот достатнє, щоб визначити природний стан білка. На підставі їх різних хімічних властивостей, деякі амінокислоти зв'язуються один з одним (наприклад, протилежно заряджені амінокислоти) і об'єднуються; інші амінокислоти прагнуть уникнути води (тому що вони є жирними), і управляють згортанням білка в таку компактну форму, яка виключає контакт води з більшістю амінокислот, які "ховаються" в ядрі цього ущільненого білка.
Чому так важко визначити природну структуру білка?
Навіть маленькі білки можуть складатися з 100 амінокислот. Число потенційних структур, доступних для навіть такого щодо маленького білка є астрономічним, тому що є дуже багато мір свободи. Обчислювати енергію кожного можливого стану (таким чином ми можемо з'ясувати, який стан є найстійкішим) - в обчислювальному відношенні важка проблема. Проблема росте по експоненті з розміром білка. Деякі людські білки можуть бути величезними (1000 амінокислот).
Як Rosetta вирішує цю проблему?
Філософія Розетти повинна використовувати і розуміння фізичних і хімічних властивостей різних типів амінокислотної взаємодії, і знання того, які локальні структури є найбільш вірогідними для коротких відрізків амінокислот прийнятими в межах білка, обмежувати область пошуку, і оцінювати енергію різних можливих структур. Перевіряючи достатньо багато структур, Розетта може знайти найнижчу енергію, найстійкішу природну структуру білка.
Чому для прогнозу структури Розетта використовує розподілені обчислення?
У багатьох випадках, де рідна структура білка вже відома, ми звернули увагу, що функція енергії Розетти може розпізнати природну структуру як стійкіше, ніж будь-який інший вибраний стан. Починаючи з випадкової форми, проте, ми відмітили, що природний стан ніколи не вибирається. Застосовуючи велику обчислювальну потужність для вирішення проблеми, ми можемо утворити ще багато форм білка, і пробовав різні стратегії пошуку побачити, яка є найефективнішою.
Як може Rosetta@home принести користь медичній науці?
Rosetta займається як дослідженнями фундаментальних методів розвитку проектування структур і комплексів білка, так і більш безпосередньо намагається боротися з такими хворобами як: Рак, ВІЧ, Малярія, Сибірська виразка, Хвороба Альцгеймера.
10. Використані джерела
Матеріали перекладені із російськомовних та англомовних джерел:
http://distributed.org.ua/index.php?go=Pages&in=cat&id=21
http://boinc.bakerlab.org/rosetta/
http://depts.washington.edu/%7Ebakerpg/publications.html
http://depts.washington.edu/%7Ebakerpg/highlights.html
http://boinc.bakerlab.org/rosetta/rah_about.php
http://www.bakerlab.org
http://predictioncenter.org
|
 ru
ru 
 |
Сайт открыт 14.12.2005
|
Сайт открыт 14.12.2005

