
Привіт Гість ( Вхід | Реєстрація )
 Розподілені обчислення в Україні > Великі проекти розподілених обчислень > World Community Grid > Завершені проекти WCG
Розподілені обчислення в Україні > Великі проекти розподілених обчислень > World Community Grid > Завершені проекти WCG
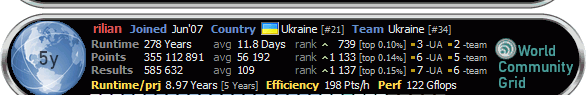
| Rilian |
 Jun 11 2008, 15:33 Jun 11 2008, 15:33
Пост
#1
|
 interstellar  Група: Team member Повідомлень: 17 200 З нами з: 22-February 06 З: Торонто Користувач №: 184 Стать: НеСкажу Free-DC_CPID Парк машин: 2 ноути і 4 компа |
 Human Proteome Folding Project Phase 2 Официальные результаты проекта Активные эксперименты Human Microbiome Project - официальный сайт http://homepages.nyu.edu/~rb133/wcg/thread_2010_03_10.html Как присоединиться читайте в главном топике World Community Grid  Proteins are essential to living beings. Just about everything in the human body involves or is made out of proteins. What are proteins? Proteins are large molecules that are made of long chains of smaller molecules called amino acids. While there are only 20 different kinds of amino acids that make up all proteins, sometimes hundreds of them make up a single protein. Adding to the complexity, proteins typically do not stay as long chains. As soon as the chain of amino acids is built, the chain folds and tangles up into a more compact and particular shape that lets it conduct specific and necessary functions within the human body. Proteins fold because the different amino acids like to stick to each other following certain rules. Imagine that amino acids are pop-beads of 20 different colors. The pop-beads are sticky, but sticky in such a way that only certain combinations of colors can stick together. This makes the amino acid chains fold in a particular way that creates proteins that are useful to the human body. Human cells have mechanisms to help the proteins fold properly and, equally important, mechanisms to get rid of improperly folded proteins. How do proteins relate to human genes? The collection of all of the human genes is known as "the human genome." Depending on how the genes are counted, there are over 30,000 genes in the human genome. Each gene, which is a section of a long chain known as DNA, dictates how to build the chain of amino acids for one of the 30,000 proteins. In recent years, scientists were able to map the sequence for each human gene. This means that we now know the sequence of amino acids in all of the human proteins. Thus, the human genome is directly related to the "human proteome," the collection of all human proteins. The protein mystery While researchers have learned a great deal about the human proteome, the functions of most of the proteins remain a mystery. The genes do not reveal exactly how the proteins will fold into their final shape, which is critical because that determines what a protein can do and what other proteins it can connect to or interact with. Proteins are like puzzle pieces. For example, muscle proteins connect to each other to form a muscle fiber. They join together in a specific manner because of their shape, as well as other factors relating to the shape. Everything that goes on in cells and in the body is very specifically controlled by the shape of the proteins that do or do not let proteins interlock with other proteins. For example, the proteins of a virus or bacteria may have particular shapes that enable it to break through the cell membrane, allowing it to infect the cell. The Human Proteome Folding Project Знания структуры белков позволит ученым понять как белки выполняют свои биологические функции, а также как болезни блокируют белки от выполнения необходимых функций для поддержания здоровых клеток The Human Proteome Folding Project will combine the power of millions of computers in a grid to help scientists understand how human proteins fold. The work to be done in this monumental task is shared across this grid, so that results can be achieved far sooner than would be possible with conventional supercomputers. With a greater understanding of protein structure, scientists can learn how diseases work and ultimately find cures for them. When your grid agent is running, it is folding an amino acid chain in various ways and evaluating how well each folding follows the specific rules of how specific amino acids stick together or not. As computers try millions of ways to fold the chains, they attempt to fold the protein in the same way that it actually folds in the human body. The best shapes identified for each protein are returned to the scientists for further study.  ----- Оказывается тут тоже юзается розетта  (Show/Hide) График проекта  Це повідомлення відредагував Rilian: Feb 4 2011, 00:23 |
   |

 
|




|
  |
Відповідей
| Rilian |
Mar 25 2010, 02:43
Пост
#2
|
|
interstellar Група: Team member Повідомлень: 17 200 З нами з: 22-February 06 З: Торонто Користувач №: 184 Стать: НеСкажу Free-DC_CPID Парк машин: 2 ноути і 4 компа |
Обновление статуса проекта за март 2010
Кратко, как я понял: изучая специальные стабильные последовательности аминокислот в предсказанных HPF2 белках, из одного поколения белка в другое (данные о белках берутся из разных популяций одного вида животных или растений), ученые проекта смотрят какие эволюционные факторы вызывают какие изменения в структуре белков, и, если возможно, какие новые функции они получают. Дальше, используя метод вероятностей (поиск по большой базе результатов, который планируется сделать в проекте с помощью мощностей WCG), ученые смогут найти 1) каким эволюционным факторам подверглись белки с неизвестными пока функциями. 2) какие функции могут приобрести белки при определенных эволюционных факторах Последнее мне кажется особенно актуальным для практического применения при создании новых генетически модифицированных организмов. Итак, статья http://homepages.nyu.edu/~rb133/wcg/thread_2010_03_10.html HPF2 Update - March 2010 Greetings WCG Volunteers, We've been working diligently to develop a pipeline for a cooperative analysis of phylogenetic and structural data. We will integrate our structure predictions with knowledge of how proteins (and functional sites on folded proteins) evolve, by estimating the phylogenies of all protein domain families in our database and identifying positively-selected amino acid sites in these families using codon-based molecular evolution models that can be mapped onto the predicted structures. The first stages of this analysis are coming to fruition, and we've begun investigating preliminary results. Using phylogenetic models, we intend to identify sites of proteins exhibiting evolutionary pressure. This may improve our understanding of how proteins evolve new functions and structures, and will ultimately lead to an increase in genome annotation for proteins whose purpose we know next to nothing about. The great scale of and wealth of information in our database may allow us to improve upon our existing and future de novo structure and function predictions. Identifying structurally or functionally importing residues in protein domains should inform our comparative modeling techniques. We use probabilistic methods to produce models of evolution using observed rates of mutation in protein families. Lots of different evolutionary pressures affect the mutation and expression of proteins, but we hope to garner insight with this analysis about how evolution adapts protein function. Using our automated methods, we produced evolutionary models for a handful of identified protein domain families in major plant genomes. One such protein family matches PDB 1TQE "Myocyte Enhancer Factor-2". While this analysis is very preliminary (and I stress preliminary), positive selection analysis identifies a few residues that may be involved in DNA binding and the integrity of the dimer near the substrate. This is the kind of science we'll be investigating in the future using WCG predicted structures. -- Patrick Winters Bonneau Lab  PDB 1TQE: the two chains colored blue and green, with probability of positive selection highlighted yellow-red. -------------------- |
|
|
|







Повідомлення у даній Темі
Rilian Human Proteonome Folding, Phase 2 Jun 11 2008, 15:33 nikelong Росетта следит за тобой!
ЗЫ: ты когда себе та... Jun 11 2008, 16:37 (_KoDAk_) :yes: Jun 11 2008, 23:33 Rilian надо бы перевести шапку...
Кратко: проект с помощ... Jan 2 2009, 16:30 Rilian Насчитал 100 процессорных дней в проекте
Это сост... Jan 4 2009, 19:27 Rilian Richard Bonneau, head scientist of the Human Prote... Jan 6 2009, 04:44 Rilian Рассчитал 500 протеинов за 142 процессорных дня Jan 11 2009, 04:16 Rilian Объявляется мини-соревнование - кто быстрее подсчи... Jan 13 2009, 21:16
nikelong Росетта следит за тобой!
ЗЫ: ты когда себе та... Jun 11 2008, 16:37 (_KoDAk_) :yes: Jun 11 2008, 23:33 Rilian надо бы перевести шапку...
Кратко: проект с помощ... Jan 2 2009, 16:30 Rilian Насчитал 100 процессорных дней в проекте
Это сост... Jan 4 2009, 19:27 Rilian Richard Bonneau, head scientist of the Human Prote... Jan 6 2009, 04:44 Rilian Рассчитал 500 протеинов за 142 процессорных дня Jan 11 2009, 04:16 Rilian Объявляется мини-соревнование - кто быстрее подсчи... Jan 13 2009, 21:16
 Rilian
Объявляется мини-соревнование - кто быстрее подсч... Mar 6 2009, 22:07 nikelong http://www.yeastrc.org/pdr/pages/search/ad...dSear... Feb 13 2009, 15:59 cosmo_vk кстати не у кого не возникало проблем с расчетом з... Mar 7 2009, 08:04 Rilian Не, но у меня на висте проблемы именно с HPFP2... Mar 7 2009, 15:23 Rilian
Привет,
Прошло уже некоторое время с тех пор ка... Mar 25 2009, 02:10 cosmo_vk А все-таки тормозятся вычисления, теперь уже такой... Apr 1 2009, 18:20 Rilian у меня на 2-гигагерцовых ксеонах бывает считает и ... Apr 1 2009, 20:50 cosmo_vk не-е у меня считает в районе 3-4 часов. Если больш... Apr 2 2009, 06:49 Rilian А... ну да, в HPFP2 оч редко такое бывает.. Может ... Apr 2 2009, 11:31 Rilian Patrick Winters продолжает радовать нас апдейтами ... Apr 8 2009, 20:54 vitalidze1 cosmo_vk,
В мене іноді такі лажі на компах вискан... May 28 2009, 16:09 cosmo_vk не-е на рисе у меня все нормально.
Глюк с этим пр... May 29 2009, 16:41
Rilian
Объявляется мини-соревнование - кто быстрее подсч... Mar 6 2009, 22:07 nikelong http://www.yeastrc.org/pdr/pages/search/ad...dSear... Feb 13 2009, 15:59 cosmo_vk кстати не у кого не возникало проблем с расчетом з... Mar 7 2009, 08:04 Rilian Не, но у меня на висте проблемы именно с HPFP2... Mar 7 2009, 15:23 Rilian
Привет,
Прошло уже некоторое время с тех пор ка... Mar 25 2009, 02:10 cosmo_vk А все-таки тормозятся вычисления, теперь уже такой... Apr 1 2009, 18:20 Rilian у меня на 2-гигагерцовых ксеонах бывает считает и ... Apr 1 2009, 20:50 cosmo_vk не-е у меня считает в районе 3-4 часов. Если больш... Apr 2 2009, 06:49 Rilian А... ну да, в HPFP2 оч редко такое бывает.. Может ... Apr 2 2009, 11:31 Rilian Patrick Winters продолжает радовать нас апдейтами ... Apr 8 2009, 20:54 vitalidze1 cosmo_vk,
В мене іноді такі лажі на компах вискан... May 28 2009, 16:09 cosmo_vk не-е на рисе у меня все нормально.
Глюк с этим пр... May 29 2009, 16:41|
|
|
1 Користувачів переглядають дану тему (1 Гостей і 0 Прихованих Користувачів)
0 Користувачів:

|
Lo-Fi Версія | Поточний час: 21st June 2026 - 14:03 |